
We recently published our latest paper in Drug Discovery Today. Our authors Fernando Garcia and Eva Gefroh introduce a type of flexible biomanufacturing facility called J.POD®, capable of capitalizing on Intensified Continuous Biomanufacturing (ICB) to deliver against current cost and speed needs in the biopharmaceutical industry. We are pleased to share this featured review with industry and partners as we continue to work together to expand access to medicines that matter.
Abstract
In this work, process models were developed to capture the impact of biomanufacturing costs on a commercial scale and emphasize the way in which facility design and operation must balance meeting product demand while minimizing production costs. Using a scenario-based modeling approach, several facility design strategies were evaluated, including a traditional large stainless-steel facility and a small footprint, portable-on-demand (POD)-based facility. Bioprocessing platforms were compared by estimating their total production costs across different facility types and specifically illustrating how continuous bioprocessing has gained in popularity as a novel and cost-effective approach to manufacture high-quality biopharmaceuticals. The analysis showed how fluctuations in market demand have a dramatic effect on manufacturing costs and plant utilization, with far-reaching implications on the total cost to patients. Read the full article in Drug Discovery Today.
Tags: Articles & Whitepapers, Biologics
A major disadvantage in fighting cancer is the low immunogenicity of cancer cells, impairing body’s ability to identify and destroy them. This paper describes STM3006, a novel, chemically distinct inhibitor of METTL3 with improved biochemical and cellular potency compared to the previously published ligand STM2457. Crystallography revealed improved binding within a newly formed ligand-induced lipophilic pocket within the protein, while retaining the interactions that provide high selectivity over other methyltransferases.
The authors demonstrated that METTL3 inhibition with STM3006 stimulates a cell-intrinsic interferon response through double-stranded RNA formation. This immunomodulatory mechanism is distinct from current immunotherapeutic agents and provides the molecular rationale for combination with anti-PD1 immune checkpoint blockade to augment anti-tumour immunity. In combination these therapies have demonstrated the ability to overcome malignant clones insensitive to the single agents.
This article is written in collaboration with Storm Therapeutics.
Tags: Articles & Whitepapers
Date: 10th -13th September 2023
Location: GR - Ljubljana Exhibition and Convention Centre, Ljubljana Slovenia
Booth Location: 59
Attending: Ben Park, Alessandro Casartelli, Oksana Nikolayenko, Daniel Gliesche, Martin Augustin, Giuseppe D'Angelo & Rob Brown
Presentations
“Genotoxicity Risk Assessment during Lead Optimization Phase in Pharmaceutical Drug Development” - Presented by Alessandro Casartelli
"Detection of Chemical Reactive Metabolites in a Glutathione depleted in vitro system" - Presented by Ben Park
Download: Bioactivation Driven Toxicity Product Sheet
Organ transplantation is often the only available treatment option for patients suffering from end-stage kidney, liver or heart disease. In collaboration with the QUOD initiative, Evotec is committed to improving the quality of organ donation and transplantation and advancing the mechanistic understanding of these common diseases. Through multimodal characterization of donor biopsy transcriptomes and associated clinical data, our scientists aim to identify early disease signatures and targetable mechanisms of injury and repair that will open new avenues for therapeutic intervention and patient stratification. For more information, please read the recent QUOD newsletter article.
Tags: Articles & Whitepapers
Blood microsampling is a less invasive and simplified alternative to traditional venipuncture for PK/TK sampling, used mainly in small-animal studies. The purpose of this work was to evaluate the possibility of using microsampling technique also to support PK/TK studies in non-human primates.
A comparison of plasma PK parameters was conducted by traditional blood collection from the femoral vein and microsampling from the tail vein of six non-naïve cynomolgus monkeys. Four drugs were selected for this comparison, based on acid-base properties and volume of distribution.
The results obtained in this work, supported by robust statistics, demonstrated the suitability of microsampling in supporting PK/TK studies in non-human primates.
The plasma exposures of the tested drugs are comparable for both sampling techniques and are not influenced by acid-base characteristics and volume of distribution.
Microsampling used in non-human primates avoids the occurrence of hematomas at the animal sampling site and can also refine practices to limit pain and distress to which animals are exposed (refinement of 3Rs) and, as a result, may reduce the impact of animal stress on PK/TK readouts; moreover, it also provides significant advantages for animal technicians during in life handling.
To request a copy of the article, contact the authors. For Evotec: massimo.breda@evotec.com
Tags: Articles & Whitepapers, Blog, ADME/DMPK, IND Enabling Studies/Preclinical Development, Toxicology & Safety
Katie Haughan, Drug Transporter Sciences Team, Cyprotex
Drug transporters play a pivotal role in drug-drug interactions (DDI’s), as such regulatory bodies such as the FDA and EMA recommend the study of specific transporters that are known to cause clinical DDI’s. One of these recommendations for orally administrated drugs, unless waivered based on the BCS classification, is the victim (substrate) potential at Breast Cancer Resistance Protein (BCRP) due to evidence that inhibition of intestinal BCRP in DDI can increase the absorption and therefore exposure of sensitive substrate drugs such as rosuvastatin and topotecan. Pharmacogenetics also need to be considered when developing drugs that are BCRP substrates as this can impact the BCRP expression levels between individuals and within the global population and can therefore result in differing pharmacokinetic profiles. Polymorphisms associated with BCRP may or may not impact on the expression or functionality of the transporter, however the prevalence of these can vary dependent on the ethnicity. One example that is of importance for understanding the pharmacogenetic impact is BCRP SNP C421A, which is associated with lower BCRP protein expression, and leads to the ABCG2-Q141K polymorphism which has a higher frequency in Japanese and Chinese populations compared to Caucasian populations [Birmingham et al. 2015, Hua et al. 2012]. As a result, the plasma levels of BCRP substrates such as rosuvastatin in these populations is increased due to greater absorption and therefore this would need to be considered for Cmax estimates and dosage strategies. Differences in BCRP expression amongst the global population can be a result of many factors such as ethnicity, genetic polymorphisms and disease states.
The industry gold standard, and regulatory recommendation, for BCRP substrate identification is the use of a polarised cell monolayer system to determine the bidirectional flux of the investigational drug in the absence and presence of a selective inhibitor. To do so there are two cell lines which are favoured across the industry; BCRP over expressing transfected Madin-Derby Canine Kidney cell line (MDCK-BCRP), or the immortalised colorectal adenocarcinoma cell line Caco-2, both of which differentiate and polarise allowing for the expression on a range of proteins and display the in vivo like characteristics such as tight junctions.
In order to select which cell line to utilise in these regulatory studies there are many aspects to consider, each cell line comes with its own advantages and disadvantages and can favour the outcome of certain classes of compounds. Whilst MDCK-BCRP has advantages over Caco-2 such as short culturing times and higher expression of the transporter of interest, there is a risk that data interpretation may be clouded due to an intrinsic limitation of the MDCK cell line which, for efflux transporter substrate determination, may result in false negatives. This potential error in the reported classification can have implications downstream as being a substrate may reduce the oral absorption and bioavailability of the drug, which may result in therapeutic dose not being achieved. Whilst transfected MDCK cells have been used for many years in order to assess an investigational drug’s BCRP substrate status, it’s applicability may be limited due to the cells lacking expression of the relevant basolateral uptake transporters that allow polar substrates to enter the cell and in turn interact with BCRP transporter on the apical membrane. Without this uptake mechanism, and in combination with the compounds poor permeability, the basolateral to apical flux would be negligible resulting in an efflux ratio (ER) less than 2 (B-A/A-B). The initial classification by the FDA is dependent on this ratio with a BCRP substrate having an efflux ratio greater than 2.
Whilst Caco-2 cells have a considerably longer culture time compared to that of MDCK cells, they have the benefit of being able to correctly identify the efflux compounds that rely upon the interplay between apical and basolateral transporters. Organic solute transporter alpha (OST-α) is expressed on the basolateral membrane of epithelial cells in the small intestine, kidney, liver, and colon amongst other organs aswell as on Caco-2 cells, and this trans-membrane protein allows passive facilitative transport of endogenous and exogenous molecules across the membrane, including polar structures. A primary function of OST-α is the transport of bile acids, and therefore lends itself to the transport of other polar substrates such as the prototypical BCRP substrates rosuvastatin and estrone 3-sulfate across the membrane and into the cell. Once inside the cell the compound then has access to the binding site of the BCRP transporter and the opportunity to be effluxed if it is indeed a substrate. For cells that lack this uptake mechanism, such as MDCK-BCRP, polar compounds have restricted entry to the cell and do not have the opportunity to be effluxed regardless of if they are a substrate of BCRP or not.
Rosuvastatin is one of the most widely prescribed statins and implicated in BCRP DDI’s due to its victim classification. Rosuvastatin is a polar compound with a logP of 0.13 and therefore has low permeability and is dependent upon uptake mechanisms to enter cells such as MDCK. The flux of rosuvastatin, apical to basolateral, basolateral to apical and the subsequent efflux ratios can be seen in Table 1 [Li et al. 2012] for three cell lines. The B-A secretory apparent permeability (and derived efflux ratio) is significantly higher (and therefore more sensitive) in Caco-2 cells compared to that in the two MDCK cell lines even though the protein expression of the BCRP transporter in Caco-2 cells would be considerably lower than in the transfected MDCK-BCRP cells, demonstrating the crucial requirement of the basolateral uptake mechanism that is present in Caco2, but absent in MDCK-BCRP, to facilitate the interaction of rosuvastatin with intracellular BCRP on the apical membrane. This factor needs to be taken into consideration when it comes to assessing the efflux potential of an investigational drug using MDCK-BCRP cells.
Table 1: Bidirectional (apical-to-basolateral (A-B) & basolateral-to-apical (B-A)) apparent permeability (Papp) and efflux ratios for rosuvastatin in different cell test systems [1]
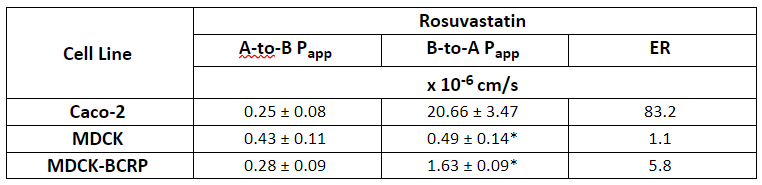
*P<0.001, significance level of the difference from the B-to-A transport in Caco-2
As shown the use of Caco-2 cells for BCRP substrate identification removes a complication seen in MDCK-BCRP cells that is dependent on the physicochemical properties of the compound, however Caco-2 cells come with their own complexities. As Caco-2 are human derived cells they express a range of endogenous human transporters including human P-glycoprotein (P-gp). BCRP and P-gp have a similar substrate profile and therefore the efflux seen in Caco-2 cells may be due to BCRP, Pgp efflux or a combination of both. In order to decipher between the two main efflux transporters a selective P-gp inhibitor such as verapamil can be added to the test system buffer to chemically knockdown P-gp activity. The residual efflux is then considered to be due to BCRP transport however this can be confirmed using a selective BCRP inhibitor, fumitremorgin C (FTC) as shown in Table 2.
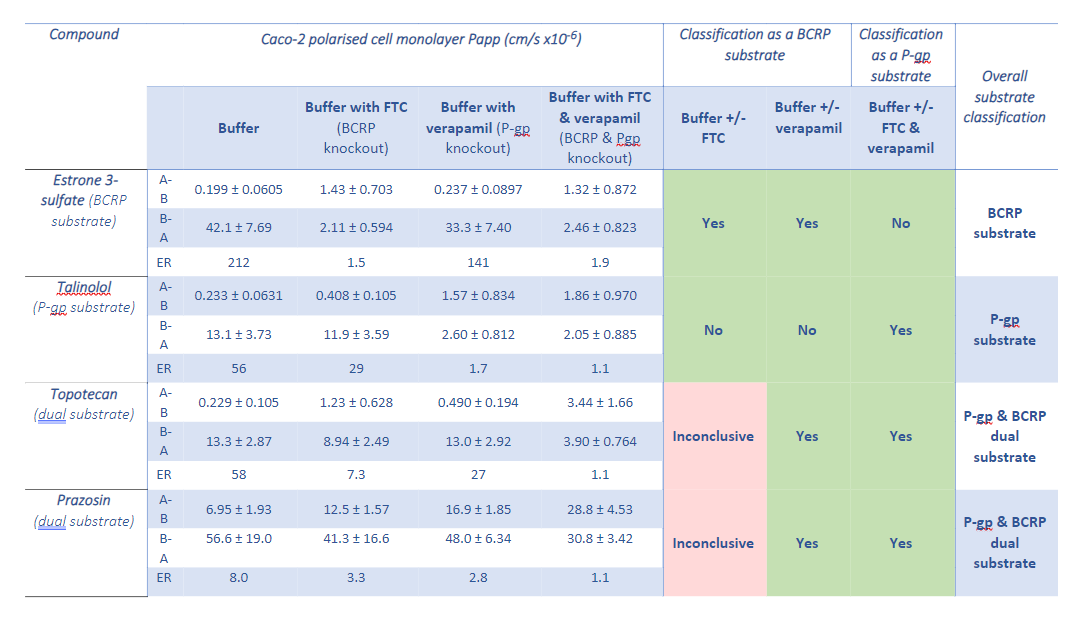
Table 2: Bidirectional (apical-to-basolateral (A-B) & basolateral-to-apical (B-A)) apparent permeability (Papp) and efflux ratios for test compounds and the substrate classifications given per assay condition
Choosing the right in vitro cell test system for BCRP substrate identification is critical for achieving the correct classification. The two gold-standard approaches have their own pros and cons, each of which can be mitigated if these test system limitations are understood. If a chemical series has a reasonable degree of lipophilicity then MDCK-BCRP should correctly identify a substrate of BCRP. However, as the industry moves towards more metabolically stable molecules and chemical series that have similar low intrinsic permeability and polarity to the clinically relevant BCRP substrate rosuvastatin, the use of Caco-2 cells would be required for the correct identification of BCRP substrates in order to avoid false negatives.
References
Rapidly growing tumor cells need a lot of oxygen and nutrients to proliferate, which requires access to the bloodstream. Therefore, tumors induce the formation of new blood vessels through a variety of molecular mechanisms. In normal tissue, there is a delicate balance of pro- and anti-angiogenic factors. In cancers, a process known as the "angiogenic switch" is initiated. As a result, pro-angiogenic signaling becomes dominant, allowing tumors to induce anarchic blood vessel formation. This switch is a critical step in the rapid growth of malignant cells, accompanied by the formation of new blood vessels.
One of the most important initiators of angiogenesis is the family of pro-angiogenic vascular endothelial growth factors (VEGF) and their receptors (VEGFR), which play an important role in both physiological and cancer angiogenesis. All members of the VEGF family stimulate cellular responses by binding to specific tyrosine kinase receptors, the VEGFRs, on the cell surface, causing them to dimerize and become activated. While VEGF-A regulates angiogenesis and vascular permeability by activating VEGFR-1 and VEGFR-2, VEGF-B seems to play a role in the maintenance of newly formed blood vessels under pathological conditions, while VEGF-C and VEGF-D and their corresponding receptor VEGFR-3 regulate lymphangiogenesis, i.e., the formation of lymphatic vessels.
This critical involvement of VEGFRs and their associated signaling pathways in the orchestration of (lymph)angiogenesis makes VEGFRs attractive targets for the treatment of tumors and the prevention of metastasis. However, existing therapies targeting VEGFRs are not very specific and inhibit a broad spectrum of receptor tyrosine kinases including the entire VEGFR family, causing many side effects such as hypertension, proteinuria, hand-foot syndrome, anorexia, and fatigue. While they show good response rates and short-term efficacy, their impact on overall survival is limited, in part because side effects limit the effective dose. This also results in only partial or transient inhibition of VEGFR-3, allowing lymphangiogenesis to serve as a tumor escape mechanism.
Evotec and Kazia therapeutics has therefore started to develop more selective VEGFR-3 receptor tyrosine kinase inhibitors. Its lead candidate, EVT801, an orally available VEGFR-3 inhibitor, is not only highly selective for VEGFR-3, but also the only inhibitor known to inhibit both VEGFR-3 homodimers and VEGFR-3:VEGFR-2 heterodimers. The compound shows low nanomolar inhibitory activity and high selectivity over kinases, various receptors, and ion channels. In November 2022, Evotec scientists reported in Cancer Research Communications that EVT801 showed potent antitumor activity in various in vitro and in vivo models. Moreover, the compound's in vivo efficacy was at least as good as that of the marketed pankinase inhibitors sorafenib and pazopanib. However, unlike sorafenib, EVT801 did not increase blood pressure in monkeys during regulatory toxicology studies or in a rat model of hypertension at doses up to 500 mg/kg, an order of magnitude higher than the pharmacological doses.
The investigators also observed that EVT801 reduced tumor (lymph)angiogenesis, apparently affecting small tumor vessels more significant than larger ones.
The efficacy of EVT801 is expected to depend on the level of VEGFR-3 expression. Interestingly, expression level of VEGFR-3 did not appear to be affected by sorafenib treatment, suggesting that EVT801 could be used in patients previously treated with any VEGFR tyrosine kinase inhibitor. However, these findings need to be confirmed in clinical trials to determine the minimum threshold of VEGFR-3 expression for effective clinical application of EVT801 and for future patient stratification.
The proposed mechanism of action of EVT801 involves three sequential anti-cancer mechanisms, all of which contribute to the inhibition of tumor growth and metastasis:
Taken together, these studies demonstrate that EVT801 is a novel anti(lymph)angiogenic agent that selectively targets VEGFR-3, modulates the tumor microenvironment to induce tumor vasculogenesis (i.e., fewer and overall larger vessels), and enhances immunotherapy.
Based on these promising results, EVT801 was selected to enter clinical trials. It is currently being evaluated as a single agent in a Phase I trial (NCT05114668) sponsored by Kazia and managed by Evotec Clinical Operations. The first stage of this Phase I study is designed as an open‑label, dose escalation trial to assess the safety, tolerability, and pharmacokinetics of EVT801 in up to 48 patients with advanced solid tumors. Details of the study were presented at the AACR Annual Meeting 2023 (Orlando, FL) (Abstract #1015). This dose escalation part will be followed by a biomarker and pharmacodynamics expansion cohort (second stage), including patients with high VEGFR-3 expressing cancers. These study sections may then be followed by a second dose escalation study, in combination with cancer immunotherapies.
The plan is to establish a patient stratification based on VEGFR-3 expression assessed by immunohistochemical imaging and immunofluorescence on tumor tissues before and after treatment. To enable this patient stratification analysis in a clinical setting, Evotec has established and validated a highly specific protocol for VEGFR-3 immunohistochemistry labeling and scoring strategy that is readily transferable to clinical sites.
To refine the VEGFR3 immunohistochemistry signature and to improve patient characterization, Evotec has developed a VEGFR3 mRNA gene signature consisting of 23 genes highly correlated with VEGFR-3 expression. The gene signature will be analyzed using the Fluidigm platform on matched FFPE patient samples. The relationship of key markers at protein and mRNA level will be investigated to potentially establish biomarkers for patient stratification and selection.
We expect that the ambitious biomarker strategy will help to better understand the effects of EVT801 in humans and may also help to select the most responsive patients and provide early indications of clinical efficacy as a monotherapy (e.g. clear renal cell carcinoma, soft tissue sarcoma and ovarian cancer) or in combination with standard of care (e.g.immune checkpoint therapies). Evotec and Kazia recently presented a scientific poster on this topic.
Hepatic transporter inhibition, mitochondrial dysfunction, and reactive metabolite formation are some of the most common mechanisms associated with intrinsic DILI. The cytochrome P450 (CYP450) superfamily of enzymes play an important role in phase 1 metabolism within the liver. For certain chemical entities, reactive metabolites may form with increased toxicity compared to the parent. These reactive metabolites may result in hepatotoxicity through the formation of reactive oxygen species, DNA damage, mitochondrial dysfunction, and endoplasmic reticulum (ER) stress. In our research the pan-specific CYP450 inhibitor, 1-aminobenzotriazole (1-ABT), was used in combination with high-content imaging to evaluate the effects of potential reactive metabolites on cell health parameters in hepatocytes. The endpoint assessed included nuclear features, glutathione (GSH) content, mitochondrial dysfunction, and reactive oxygen species (ROS) formation, as well as cellular ATP content. A calculated fold-shift in cell health features between the plus and minus 1-ABT dosing conditions was used to determine reactive metabolite formation. A panel of known DILI reference compounds associated with the formation of reactive metabolites were assessed through this HCI bioactivation assay within metabolically competent HepaRG cells, primary human hepatocytes (PHH) and primary mouse hepatocyte (PMH).
Tags: Posters, Toxicology & Safety
Peripheral neuropathy can be induced by many chemotherapeutics. Symptoms include numbness, tingling or abnormal sensations which can impact on the long-term quality of a patient’s life. Animal models used to evaluate these side effects may be difficult to interpret and are labor-intensive. In this study, we further optimized a previously developed DRG assay by comparing the neurite outgrowth responses of a group of chemotherapeutics from different classes at 24 h and 72 as an in vitro cell-based model for peripheral neuropathy. Cytotoxicity was assessed alongside neurite outgrowth. Taxanes (paclitaxel, docetaxel), microtubule interfering agents (vincristine, vinblastine, colchicine, nocodazole) and epothilones (ixabepilone) were assessed.
Tags: Posters, Toxicology & Safety