Presentation slide deck for the webinar Deciphering the Clinincal DDI Between Atazanavir and Rosuvastatin.

Presentation slide deck for the webinar Deciphering the Clinincal DDI Between Atazanavir and Rosuvastatin.
Tags: Presentations, ADME/DMPK
Rapid turnaround is essential for ADME-Tox services, especially during drug discovery. However, turnaround time for the assay isn’t the only consideration. The time required to prepare the quotation may delay the scheduling of the assay and, in turn, the study start date.
The new Cyprotex e-Store provides a solution to this. Registered users of the e-Store are able to view instant pricing for a comprehensive range of ADME-Tox services. As well as having access to detailed protocols and the latest editions of our educational guides, users also benefit from special offers only available through the e-Store.
Online ordering is simple. Previous orders can be tracked through the e-Store and it is easy to re-order your favourite assays. All of this information can be accessed outside business hours. A range of payment options are available on the e-Store including credit card, existing purchase order or invoice. These features allow services to be ordered and scheduled the same day so saving you time.
Despite this automated approach, you are still supported by a dedicated scientific study manager and account manager who are on-hand to guide you at all stages of your study.
Want to learn more?
The industry gold-standard in vitro cell based test system for studying Breast Cancer Resistance Protein (BCRP) or P-glycoprotein (P-gp) transporters is the polarized cell monolayer system using either Caco-2 cells or MDCK-MDR1 cells (Elsby et al., 2022), grown on semi permeable membrane inserts to form a brush border membrane barrier separating two experimental compartments (apical and basolateral) of equivalent pH (7.4). Apparent permeability (Papp, cm/s x10-6) of the test article is determined in the apical to basolateral (A-B) and the basolateral to apical (B-A) direction and from this an efflux ratio is calculated (B-A Papp/A-B Papp). An efflux ratio greater than 2 which is reduced by greater than 50% with a corresponding reduction in B-A Papp in the presence of a reference inhibitor (Atkinson et al., 2022) indicates the test article is a substrate of the efflux transporter being investigated. The bidirectional Papp values and subsequent efflux ratios of the known P-gp substrate quinidine, determined in polarized MDCK-MDR1 cell monolayers across a concentration range is summarized in Table 1 (left) and demonstrates that in the MDCK-MDR1 polarized monolayer test system quinidine has been determined to be a substrate of the P-gp transporter.
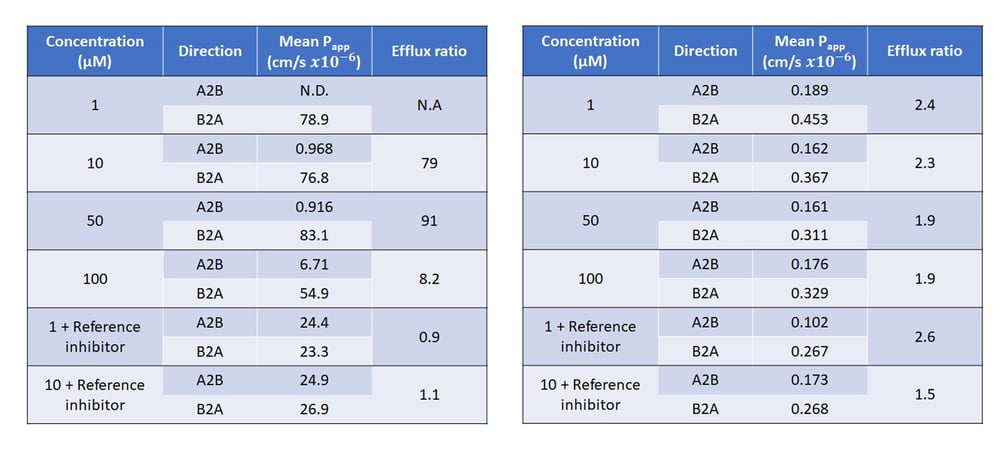
Table 1: Mean Papp values and corresponding efflux ratios of quinidine (left) and N-methyl quinidine (right) across MDCK-MDR1 monolayer cells in the presence and absence of reference inhibitor cyclosporin A
Despite polarized monolayers being the industry gold standard test system for studying P-gp and BCRP interactions, there are limitations and risk of inconclusive results and false negatives if the test article being studied exhibits low Papp indicating inherently low passive membrane permeability. This is due to the physico-chemical characteristics of the test article limiting cellular entry required to access the efflux transporter on the apical membrane. This is true for the quinidine oxidative metabolite, N-methyl quinidine (NMQ). The presence of the methyl group results in lower lipophilicity and therefore passive permeability without diminishing its ability to bind P-gp. As summarized in Table 1 (right) bidirectional Papp values for the more polar NMQ are very low and although efflux ratios determined are around two, a 50% reduction in efflux ratio with corresponding decrease in B-A Papp is not observed in the presence of inhibitor therefore the result is inconclusive at best, or at worst could be interpreted as not a substrate of the P-gp transporter.
Where assessment of a test article in polarized monolayer cells indicates a compound has inherently low passive permeability, membrane vesicles have the membrane orientated “inside out”, this results in the intracellular binding site of the transporter being positioned on the outside of the vesicle (outwardly facing), therefore compounds do not need to cross a membrane in order to interact with the transporter. As such for efflux transporters such as P-gp or BCRP, substrates will be actively taken up into the vesicle and, due to poor permeability, will remain inside the vesicle for quantification. As demonstrated in Table 2, using P-gp membrane vesicles as an in vitro test system for P-gp substrate identification, NMQ is clearly demonstrated to be, and subsequently correctly identified as, a substrate of the P-gp transporter.

Table 2: Mean uptake rate and corresponding uptake ratios of N-methyl quinidine using P-gp expressing membrane vesicles in the presence of ATP or AMP in the absence and presence of the reference inhibitor cyclosporin A. Figure: Mean uptake rate of N-methyl quinidine into P-gp expressing membrane vesicles in the presence of ATP or AMP in the absence and presence of the reference inhibitor (I) cyclosporin A
Furthermore, if data from P-gp or BCRP substrate identification studies in polarized cell monolayers indicate a compound has inherently low passive permeability then any P-gp or BCRP inhibition data generated in the same in vitro test system should also be scrutinized and a follow up inhibition study in membrane vesicles considered, particularly if no inhibition was observed in the cells.
Whilst membrane vesicles have their advantages over polarized cell monolayers as a test system to identify P-gp or BCRP substrates that are poorly permeable, it is not advisable to utilize membrane vesicles as a first-choice test system. This is because for lipophilic test articles such as quinidine, sufficient passive permeability allows the substrate to move freely across the membrane and would not be trapped within the vesicle lumen, therefore there is a risk of inconclusive results and false negatives.
In summary, polarized cell monolayers are the gold standard for studying P-gp and BCRP interactions as indicated in regulatory guidances and as such should be the first choice of test system for identifying P-gp or BCRP substrates and inhibitors. However, if results indicate the compound has inherently low passive permeability then P-gp or BCRP expressing membrane vesicles should be considered as an alternative follow up in vitro test system to mitigate against the clinical implications of false negatives.
References:
Atkinson, H., Mahon-Smith, K. and Elsby, R. (2022) ‘Drug permeability and transporter assessment: Polarized Cell Lines’, The ADME Encyclopedia, pp. 401–412. doi:10.1007/978-3-030-84860-6_142.
Elsby, R. et al. (2022) Studying the right transporter at the right time: An in vitro strategy for assessing drug-drug interaction risk during drug discovery and development. Expert Opin Drug Metab Toxicol 18(10):. 619–655. doi:10.1080/17425255.2022.2132932.
This week we celebrate Mitochondrial Awareness Week, a dedicated time to turn our attention to these tiny yet incredibly powerful cellular structures that play a pivotal role in our health and well-being. While the mention of mitochondria might not immediately conjure up images of groundbreaking scientific discoveries, these dynamic organelles deserve a moment in the spotlight. They are the unsung heroes of our cells, and their importance extends far beyond just being the "powerhouses" that produce cellular energy.
In addition to being cellular “powerhouses” and generating the majority of a cells energy in the form of adenosine triphosphate (ATP), mitochondria are also involvement in apoptosis, calcium signalling, regulation of cellular metabolism and proliferation of haem and steroids.
Unfortunately, mitochondria are also a common target for drug-induced toxicity which can have a significant adverse effect on cellular function and overall health. Here are some key reasons why understanding the potential for mitochondrial toxicity is crucial when screening for a drug candidate:
In summary, studying mitochondrial toxicity is essential to ensure the safety and efficacy of drug candidates. It helps prevent potential harm to patients, reduces the risk of adverse effects, and supports the development of drugs that are not only effective but also safe for long-term use.
Cyprotex offer a number of approaches to study mitochondrial toxicity, including:
In addition, we frequently publish our work in the field of understanding mitochondrial toxicity. You may be interested in:
Publication: A Combined In Vitro Approach to Improve the Prediction of Mitochondrial Toxicants
Contact us to find out more about our mitochondrial toxicity services today!
Blood microsampling is a less invasive and simplified alternative to traditional venipuncture for PK/TK sampling, used mainly in small-animal studies. The purpose of this work was to evaluate the possibility of using microsampling technique also to support PK/TK studies in non-human primates.
A comparison of plasma PK parameters was conducted by traditional blood collection from the femoral vein and microsampling from the tail vein of six non-naïve cynomolgus monkeys. Four drugs were selected for this comparison, based on acid-base properties and volume of distribution.
The results obtained in this work, supported by robust statistics, demonstrated the suitability of microsampling in supporting PK/TK studies in non-human primates.
The plasma exposures of the tested drugs are comparable for both sampling techniques and are not influenced by acid-base characteristics and volume of distribution.
Microsampling used in non-human primates avoids the occurrence of hematomas at the animal sampling site and can also refine practices to limit pain and distress to which animals are exposed (refinement of 3Rs) and, as a result, may reduce the impact of animal stress on PK/TK readouts; moreover, it also provides significant advantages for animal technicians during in life handling.
To request a copy of the article, contact the authors. For Evotec: massimo.breda@evotec.com
Tags: Articles & Whitepapers, Blog, ADME/DMPK, IND Enabling Studies/Preclinical Development, Toxicology & Safety
Katie Haughan, Drug Transporter Sciences Team, Cyprotex
Drug transporters play a pivotal role in drug-drug interactions (DDI’s), as such regulatory bodies such as the FDA and EMA recommend the study of specific transporters that are known to cause clinical DDI’s. One of these recommendations for orally administrated drugs, unless waivered based on the BCS classification, is the victim (substrate) potential at Breast Cancer Resistance Protein (BCRP) due to evidence that inhibition of intestinal BCRP in DDI can increase the absorption and therefore exposure of sensitive substrate drugs such as rosuvastatin and topotecan. Pharmacogenetics also need to be considered when developing drugs that are BCRP substrates as this can impact the BCRP expression levels between individuals and within the global population and can therefore result in differing pharmacokinetic profiles. Polymorphisms associated with BCRP may or may not impact on the expression or functionality of the transporter, however the prevalence of these can vary dependent on the ethnicity. One example that is of importance for understanding the pharmacogenetic impact is BCRP SNP C421A, which is associated with lower BCRP protein expression, and leads to the ABCG2-Q141K polymorphism which has a higher frequency in Japanese and Chinese populations compared to Caucasian populations [Birmingham et al. 2015, Hua et al. 2012]. As a result, the plasma levels of BCRP substrates such as rosuvastatin in these populations is increased due to greater absorption and therefore this would need to be considered for Cmax estimates and dosage strategies. Differences in BCRP expression amongst the global population can be a result of many factors such as ethnicity, genetic polymorphisms and disease states.
The industry gold standard, and regulatory recommendation, for BCRP substrate identification is the use of a polarised cell monolayer system to determine the bidirectional flux of the investigational drug in the absence and presence of a selective inhibitor. To do so there are two cell lines which are favoured across the industry; BCRP over expressing transfected Madin-Derby Canine Kidney cell line (MDCK-BCRP), or the immortalised colorectal adenocarcinoma cell line Caco-2, both of which differentiate and polarise allowing for the expression on a range of proteins and display the in vivo like characteristics such as tight junctions.
In order to select which cell line to utilise in these regulatory studies there are many aspects to consider, each cell line comes with its own advantages and disadvantages and can favour the outcome of certain classes of compounds. Whilst MDCK-BCRP has advantages over Caco-2 such as short culturing times and higher expression of the transporter of interest, there is a risk that data interpretation may be clouded due to an intrinsic limitation of the MDCK cell line which, for efflux transporter substrate determination, may result in false negatives. This potential error in the reported classification can have implications downstream as being a substrate may reduce the oral absorption and bioavailability of the drug, which may result in therapeutic dose not being achieved. Whilst transfected MDCK cells have been used for many years in order to assess an investigational drug’s BCRP substrate status, it’s applicability may be limited due to the cells lacking expression of the relevant basolateral uptake transporters that allow polar substrates to enter the cell and in turn interact with BCRP transporter on the apical membrane. Without this uptake mechanism, and in combination with the compounds poor permeability, the basolateral to apical flux would be negligible resulting in an efflux ratio (ER) less than 2 (B-A/A-B). The initial classification by the FDA is dependent on this ratio with a BCRP substrate having an efflux ratio greater than 2.
Whilst Caco-2 cells have a considerably longer culture time compared to that of MDCK cells, they have the benefit of being able to correctly identify the efflux compounds that rely upon the interplay between apical and basolateral transporters. Organic solute transporter alpha (OST-α) is expressed on the basolateral membrane of epithelial cells in the small intestine, kidney, liver, and colon amongst other organs aswell as on Caco-2 cells, and this trans-membrane protein allows passive facilitative transport of endogenous and exogenous molecules across the membrane, including polar structures. A primary function of OST-α is the transport of bile acids, and therefore lends itself to the transport of other polar substrates such as the prototypical BCRP substrates rosuvastatin and estrone 3-sulfate across the membrane and into the cell. Once inside the cell the compound then has access to the binding site of the BCRP transporter and the opportunity to be effluxed if it is indeed a substrate. For cells that lack this uptake mechanism, such as MDCK-BCRP, polar compounds have restricted entry to the cell and do not have the opportunity to be effluxed regardless of if they are a substrate of BCRP or not.
Rosuvastatin is one of the most widely prescribed statins and implicated in BCRP DDI’s due to its victim classification. Rosuvastatin is a polar compound with a logP of 0.13 and therefore has low permeability and is dependent upon uptake mechanisms to enter cells such as MDCK. The flux of rosuvastatin, apical to basolateral, basolateral to apical and the subsequent efflux ratios can be seen in Table 1 [Li et al. 2012] for three cell lines. The B-A secretory apparent permeability (and derived efflux ratio) is significantly higher (and therefore more sensitive) in Caco-2 cells compared to that in the two MDCK cell lines even though the protein expression of the BCRP transporter in Caco-2 cells would be considerably lower than in the transfected MDCK-BCRP cells, demonstrating the crucial requirement of the basolateral uptake mechanism that is present in Caco2, but absent in MDCK-BCRP, to facilitate the interaction of rosuvastatin with intracellular BCRP on the apical membrane. This factor needs to be taken into consideration when it comes to assessing the efflux potential of an investigational drug using MDCK-BCRP cells.
Table 1: Bidirectional (apical-to-basolateral (A-B) & basolateral-to-apical (B-A)) apparent permeability (Papp) and efflux ratios for rosuvastatin in different cell test systems [1]
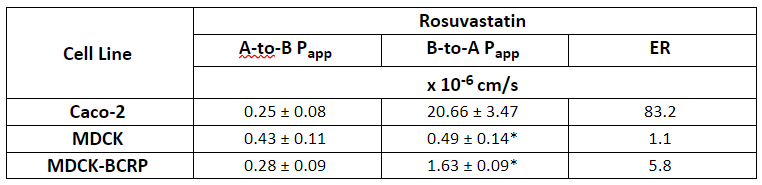
*P<0.001, significance level of the difference from the B-to-A transport in Caco-2
As shown the use of Caco-2 cells for BCRP substrate identification removes a complication seen in MDCK-BCRP cells that is dependent on the physicochemical properties of the compound, however Caco-2 cells come with their own complexities. As Caco-2 are human derived cells they express a range of endogenous human transporters including human P-glycoprotein (P-gp). BCRP and P-gp have a similar substrate profile and therefore the efflux seen in Caco-2 cells may be due to BCRP, Pgp efflux or a combination of both. In order to decipher between the two main efflux transporters a selective P-gp inhibitor such as verapamil can be added to the test system buffer to chemically knockdown P-gp activity. The residual efflux is then considered to be due to BCRP transport however this can be confirmed using a selective BCRP inhibitor, fumitremorgin C (FTC) as shown in Table 2.
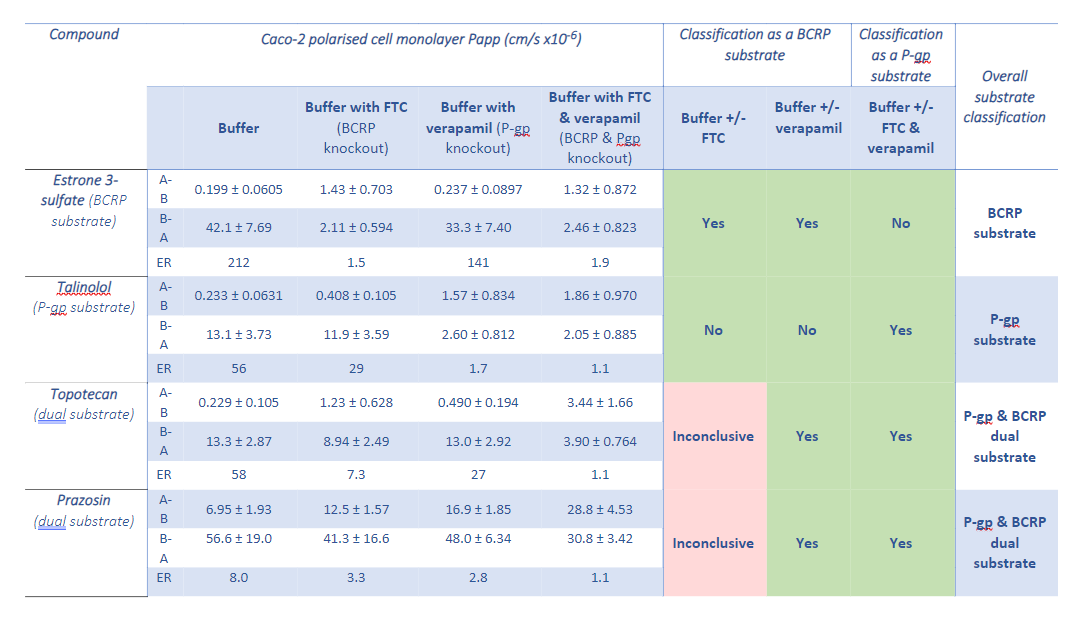
Table 2: Bidirectional (apical-to-basolateral (A-B) & basolateral-to-apical (B-A)) apparent permeability (Papp) and efflux ratios for test compounds and the substrate classifications given per assay condition
Choosing the right in vitro cell test system for BCRP substrate identification is critical for achieving the correct classification. The two gold-standard approaches have their own pros and cons, each of which can be mitigated if these test system limitations are understood. If a chemical series has a reasonable degree of lipophilicity then MDCK-BCRP should correctly identify a substrate of BCRP. However, as the industry moves towards more metabolically stable molecules and chemical series that have similar low intrinsic permeability and polarity to the clinically relevant BCRP substrate rosuvastatin, the use of Caco-2 cells would be required for the correct identification of BCRP substrates in order to avoid false negatives.
References
Our Drug Transporter Sciences team at Cyprotex are delighted to announce publication of a review article in Expert Opinion on Drug Metabolism and Toxicology entitled ‘Studying the Right Transporter at the Right Time: An In Vitro Strategy for Assessing Drug-Drug Interaction Risk during Drug Discovery and Development’.
As our understanding of the role of drug transporters in clinical drug-drug interactions (DDIs) has developed, the list of transporters requiring in vitro study by regulators has grown to accommodate assessment of risk for new drugs. Currently, ten transporters require routine study prior to regulatory NDA submission. Getting the timing wrong for these investigations could result in in vitro data being generated either 1) too early in the drug discovery/development timeline and potentially becoming surplus to requirements if the investigational drug fails for reasons of poor pharmacokinetics (and efficacy) or toxicity, or 2) too late to influence finalisation of the clinical development plan resulting in perhaps unnecessary comedication exclusions that impact patient recruitment and thus delay clinical trials. In either case, there will be a cost and resource penalty, with the overall impact being considerably cheaper for the former compared with the latter. To minimize these development risks, project teams should study the right transporters at the right time for their investigational drug and the authors (Dr’s Robert Elsby, Hayley Atkinson, Philip Butler and Rob Riley) have tried to address this in their review article by proposing in vitro strategies that could be employed to either mitigate/remove transporter DDI risk during development through frontloading certain studies, or to manage (contextualize) DDI risk to patients in the clinical setting.
In the article, an overview of clinically relevant drug transporters and observed DDIs is provided, alongside presentation of key considerations/recommendations for in vitro study design when evaluating drugs as inhibitors or substrates of transporters. Guidance on identifying critical victim comedications and their clinically relevant disposition pathways, and using mechanistic static equations for quantitative prediction of DDI (demonstrating a 97% predictive accuracy for 28 statin DDIs) is also compiled. To truly alleviate or manage clinical risk, the industry would benefit from moving away from current regulatory qualitative basic static equation approaches to quantitative mechanistic DDI prediction, thereby contextualising risk to ascertain whether a transporter DDI is simply pharmacokinetic or clinically significant requiring intervention. Furthermore, such a mechanistic approach can be used towards either mitigating perpetrator DDI risk early during candidate selection, or managing clinical risk and aiding patient recruitment by informing labels and potentially providing an alternative to conducting costly clinical interaction studies with co-medications in the future.
Cytochrome (CYP) P450 induction is a well-established mechanism for increased prevalence of DDIs. Whilst its study in vitro has been described in regulatory guidance for over 20 years, data interpretation methods and details for study design have evolved and the best approaches for study of small molecules and other therapeutics are still being discussed within CROs, pharma and biotechs.
In this webinar our expert, Katie Plant, describes the most recent updates in CYP induction. By reviewing and consolidating literature and recent perspectives from Innovation and Quality Induction Working Group (IQ IWG) alongside the regulatory guidance, this provides a comprehensive overview and outlines the recommended approach to assess CYP induction in vitro. When supported by well-designed in vitro assays and batch qualification using a known set of CYP3A4 inducers, relative induction score (RIS) correlation analysis serves as a useful tool enabling the magnitude of clinical CYP3A4 induction to be predicted and used to support decision-making for investigating potential DDI risk.
 |
Katie Plant Principal Scientist | Cyprotex Discovery Ltd Katie Plant is a Principal Scientist in the Drug Metabolism group at Cyprotex, UK, responsible for operational delivery of high-throughput screening assays covering metabolic stability, enzyme inhibition and induction, alongside supporting clients in designing and interpreting DDI studies to support regulatory submission, supporting R&D and bespoke studies and driving continuous improvement projects. |
Watch the webinar to learn more!
[hs-form id="f0365c7a-f5d3-411a-a11b-097b618db7a5" show="hide on login" url="https://news.evotecsciencepool.com/hubfs/Sciencepool/Videos%20and%20Webinars/Cyprotex%20webinars%20and%20videos/cyp-induction-webcast-on-demand-july2022.mp4"]
Tags: Videos & Webinars, ADME/DMPK
Toxicokinetic evaluation is a regulatory and scientific requirement in the drug development process. To obtain plasma, blood is generally withdrawn by a conventional venous collection method. Microsampling is a less invasive sampling technique, which allows to reduce the stress correlated to the conventional blood sampling and to decrease the number of rodents for a preclinical study. The implementation of microsampling in particular, in non-human primate can reduce the stress and promote a positive interaction with technical staff which improves the overall well-being of the animal (refinement).
In this poster we summarise the work done to evaluate the possible influence of the blood sampling method on drug plasma concentrations, using LC-MS/MS methods in non-human primate for four drugs selected based on acid-base and volume of distribution properties.
The poster was presented by our expert Rossella Cardin at the 24th International Reid Bioanalytical Forum held in Cambridge, UK, on June 13-16, 2022.
Tags: Posters, ADME/DMPK, IND Enabling Studies/Preclinical Development, Toxicology & Safety
Tags: Fact Sheets, Formulation & CMC, ADME/DMPK, IND Enabling Studies/Preclinical Development, Toxicology & Safety