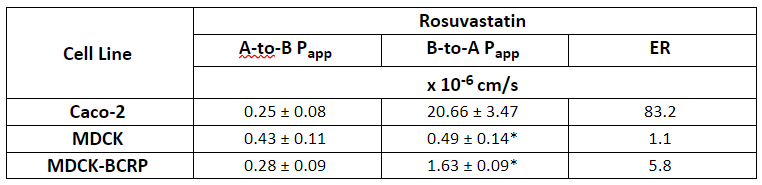
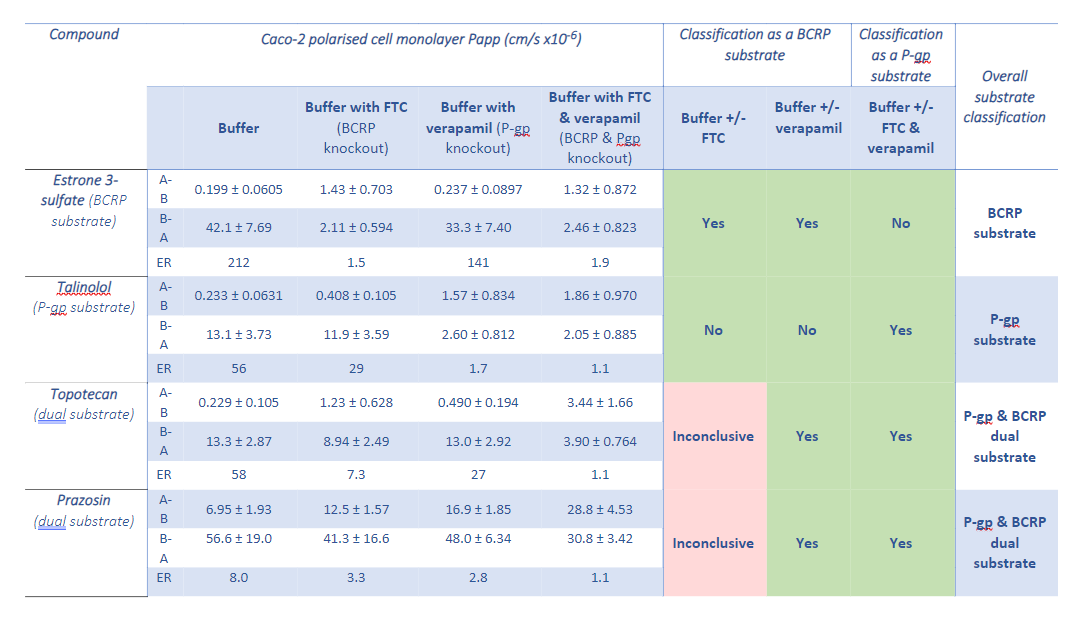
According to a study from 2020, a total of 133 drugs were withdrawn from the market due to safety reasons between 1990 and 2010. Major causes were hepatotoxicity (27.1%), cardiac disorders (18.8%), hypersensitivity (12.8%), and nephrotoxicity (9.8%), accounting for 69.2% of all drugs withdrawn. In most cases, these withdrawals were initiated because of spontaneous reports and/or case reports. Another study looking into drug withdrawals between 1953 and 2013 revealed that 18% of drug withdrawals from the market in this period were due to liver damage.
Add to these withdrawals of marketed drugs the attrition rate of drug candidates in clinical trials: 90 percent of all drug candidates fail in clinical trials, and 30 percent of these failures are due to unmanageable toxicity issues.
These failures occur despite thorough preclinical work and intensive animal studies. It is estimated that only 50% of the compounds that cause liver toxicity in humans are detected by animal studies. Furthermore, some adverse reactions or idiosyncratic toxic effects are typically not detected until the drug in question has gained large exposure in a broad patient population.
Interestingly, a study evaluating the attrition of drug candidates from AstraZeneca, Eli Lilly and Company, GlaxoSmithKline and Pfizer came to the conclusion that there is a strong link between physicochemical properties of compounds and clinical failure due to safety issues. The results also suggest that further control of physicochemical properties is unlikely to have a significant effect on attrition rates and that additional work is required to address safety-related failures.
These failures are not only costly (according to the FDA, drug development takes over 10–15 years with an average cost of over $1–2 billion for each new drug to be approved), but are also putting the health and the life of patients in danger.
Consequently, Cyprotex and its parent company Evotec are very focused on assessing toxicology issues from the very beginning of its drug R&D process and have invested a significant amount of time and resources to expand its technologies for the toxicological evaluation of drug candidates.
“The idea is to make better informed decisions earlier in your discovery campaign when you can select potentially safer compounds, rather than finding a safety liability later on,” says Paul Walker PhD, Vice President, Head of Toxicology at Cyprotex, in Cheshire, UK.
This improved discovery and selection is implemented by Cyprotex by using the unbiased view of transcriptomics and its potential to predict drug-induced toxicity. Transcriptomics involves sequencing thousands of mRNA molecules to identify which processes are active in the cell and allows for a better understanding of the cell’s reaction to known and novel drugs.
This is by no means a purely academic endeavour. As an example, the Cyprotex team demonstrated via transcriptomics it was able to identify problems in liver cells treated with fasiglifam, a promising diabetes drug candidate, which was withdrawn from late-stage clinical trials by its developer, following signs of liver damage in trial participants. This example proves that transcriptomics could have raised a red flag during preclinical development and might have saved hundreds of millions of dollars.
“Our studies have found potential effects on mitochondrial function, which were previously missed in preclinical studies” says Walker.
Therefore, transcriptomics has the potential to supplement or reduce in vivo toxicology studies by effectively identifying safety issues early in drug development, saving time and money — and animal testing.
Sophisticated Human Cell-Based Models
A key advantage of transcriptomics is its use of human cells and Evotec as well as Cyprotex are not just looking at 2D cell cultures, but investigating 3D organoids. These structures formed of thousands of cells that mimic organ-specific tissues are much closer to the real organ and have valuable features: For example, 3D-organoids of the heart exhibit regular contractions, beating like a living heart, and liver organoids secrete typical liver enzymes for days.
“On top of that, a 3D system allows repeat dosing, mimicking dosing regimens in vivo and potentially helps to detect effects due to toxic metabolites,” says Walker.
As they are small, the organoids can be placed in 384-well plates and individually molecular barcoded for simultaneous sequencing. This combination of miniaturization and high-throughput screening is implemented in Evotec’s EVOpanOmics platform and allows a wider adoption of transcriptomics in preclinical toxicology studies allowing for the repeat testing of dozens or even hundreds of compounds at several doses and in multiple organs.
“People have thought about using transcriptomics for toxicology before, but it was always a numbers game,” explains Rüdiger Fritsch PhD, Principal Scientist and Project Lead for EVOpanOmics. “For any compound that’s a real troublemaker, the evidence will show up in the transcriptomics data if you profile it in a relevant model. You just need to test appropriate dosing scenarios with the breadth of genome-wide off-target effects so that you have a chance to find it.”
Complex Analysis of Transcriptomics Data
Evotec, in conjunction with Cyprotex, offers transcriptomics services to drug developers and carries out the entire process in-house, from growing the organoids to sequencing and analysis. This streamlined process allows its researchers to screen hundreds of compounds a day, each delivering tens of thousands of data points on RNA levels. To analyze all of these vast amounts of data, Evotec has developed a software platform called EVOpanHunter that allows among others the analysis of these transcriptomics in an interactive manner.
“We want to democratize data analysis for the biologists who know the biological pathways and processes, without them needing to rely on additional experts from the bioinformatics department for routine tasks,” says Carla Tameling PhD, Head of Sales and Application for EVOpanHunter at Evotec.
On top of the interactive multi-omics analysis platform machine learning is used to trawl through this immense amount of data in order to find specific patterns hinting for toxicological effects and alert the researchers to dig deeper. “The more data we get, the harder it is for a human to dig through it all,” adds Tameling. “Transcriptomics is an unbiased view. You don’t need to define what to look at prior to your studies — you get all the data, and you might see things that you didn’t think would be relevant initially.”
From publically available sources, Cyprotex has compiled a broad and highly valuable transcriptomics reference database for drug-induced liver injuries.. Machine learning is being applied to predict whether a compound is likely to have issues by comparing the observed pattern of gene activity to the activity patterns of known toxic molecules. Furthermore, this is not restricted to hepotoxicity. Cyprotex is already building databases of other organs, such as heart, kidney and brain, using publicly available drug development trial results to select a broad space of reference comounds. “We’re running reference compounds from all kinds of sources where we know there are either late-stage clinical findings or withdrawals from the market,” states Walker.
Given the rapid advancements of the technology, it may be only a matter of time before transcriptomics and other omics technologies become a regulatory standard approach for preclinical toxicity testing.
Contact Us for More Information
Learn More